Biochemical and Biophysical Systems Group
The Biochemical and Biophysical Systems Group’s scientists use cutting-edge, multi-scale, in silico simulations to tackle problems in biology. We use a wide range of computational biology methods that employ LLNL’s high-performance computing resources to simulate systems from sub-atomic scale to population level. These methods include atomistic and coarse-grained molecular dynamics, quantum simulations, constraint-based genome-scale simulations, reaction-transport dynamic simulations, and agent-based, whole-organ, and pharmacokinetics/pharmacodynamics models. We develop new computational methods to describe and predict biological systems. In addition, we combine experimental efforts with physics-based simulations and statistical and machine-learning models to accelerate the design and development of safe and effective therapeutics. Overall, we seek predictive understanding of protein-mediated processes related to critical missions of LLNL, including bioenergy, medical countermeasures, and new materials.
Research Area Highlights
-
Fighting cancer: behavior of mutant proteins simulated at unprecedented length- and time-scales
-
Using supercomputers to search for small-molecule inhibitors of SARS-CoV-2 proteins
-
Research partnership with the American Heart Association
-
An electrostatic funnel in the GABA-binding pathway
-
Drug development using high-performance computing for a new class of antibiotics

Fighting cancer: behavior of mutant proteins simulated at unprecedented length- and time-scales
Contact: Helgi Ingolfsson
RAS is a signaling protein associated with the cell membrane that is mutated in up to 30% of human cancers. RAS signaling has been proposed to be regulated by the dynamic heterogeneity of the cell membrane, in particular, the distribution/density of different types of lipids potentially affects RAS co-localization and dimerization.
Investigating such effects requires simulation at near-atomistic detail to be extended to macroscopic temporal and spatial scales, which is not possible with conventional computational or experimental techniques. BBS and collaborators developed a computational framework -- the Multiscale Machine-Learned Modeling Infrastructure (MuMMI) -- that uses machine learning to create a scale-bridging ensemble of over 100,000 simulations of active wild-type RAS on a complex, asymmetric membrane. Initialized and validated with experimental data (including a new crystal structure of active wild-type RAS), these simulations represent a substantial advance in the ability to characterize RAS-membrane biology.
We found distinctive patterns of local lipid composition that correlate with interfacially promiscuous RAS multimerization. These lipid fingerprints are coupled to RAS dynamics, predicted to influence effector binding, and therefore may be a mechanism for regulating cell signaling cascades.
These figures outline the overall approach, which uses experimental results to initialize the computations.
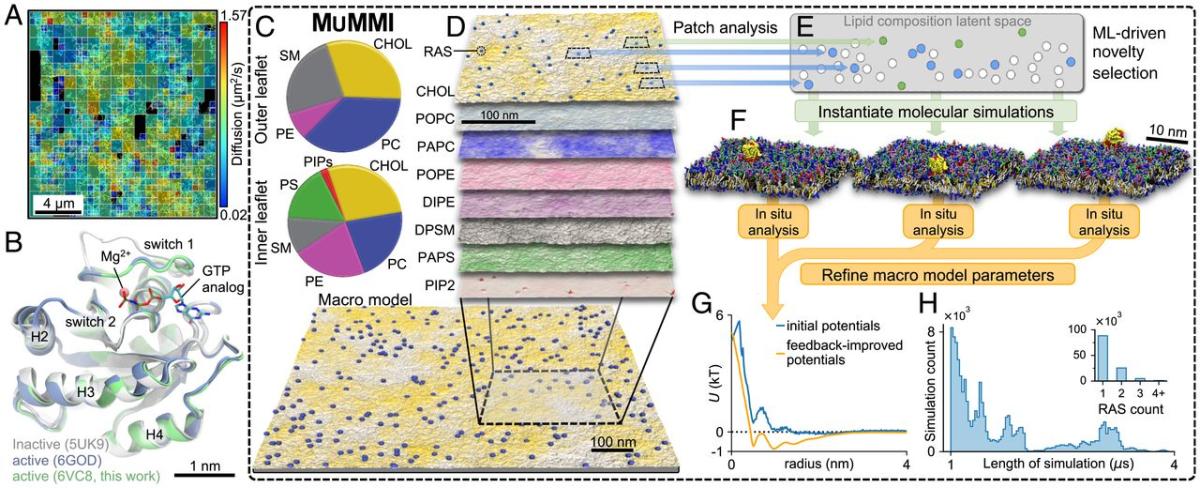
Figure (A) shows the experimentally determined diffusion mapping of single molecules of RAS tethered to or within 100 nm of the cell membrane in a 16 × 16-μm2 region of a live HeLa cell accumulated over 10 s. Figure (B) represents crystal structures of wild-type RAS in active (green and blue) and inactive (gray) configurations. The pie charts in figure (C) show average macro model lipid composition in the outer portion of the lipid bilayer (top) and the inner portion (bottom). Figures (D-F) illustrate the Multiscale Machine-learned Modeling Infrastructure (MuMMI). Figure (D) shows representative snapshots of each of the different lipid distributions in the inner leaflet of a 0.3 × 0.3-μm2 region of the full 1 × 1-μm2 macro simulation; color saturation indicates local lipid density. Figure (E) presents a schematic of the latent space encoding of lipid composition in 30 × 30-nm2 membrane patches. From the candidate patches (blue and green), those that are most dissimilar (green) to existing (white) near-atomistic scale (coarse-grained) simulations are selected and used to spawn new coarse-grained simulations. Figure (F) shows representative coarse-grained simulation systems (water molecules are not shown). Figure (G) shows the improvement of macro model parameter inputs from feedback iteration. Figure (H) shows the distribution of coarse-grained simulation duration and (Inset) the number of RAS proteins per membrane patch.
LLNL summary/release: Unprecedented multiscale model of protein behavior linked to cancer-causing mutations
Supporting data: https://bbs-int.llnl.gov/RAS-lipid-dependent-dynamics-data
Using supercomputers to search for small-molecule inhibitors of SARS-CoV-2 proteins
Contact: Ed Lau
The COVID-19 pandemic, and the lack of effective treatments, poses a real-world challenge for drug discovery. We used supercomputers to search for molecules that are predicted to bind to two SARS-CoV-2 protein targets – potentially inhibiting the virus. The proteins are the “main protease,” essential to viral replication, and the “spike” protein, required to invade human cells. We used “docking” software to fit over 26 million molecules, including 5400 existing drugs, into pockets on the surface of each of these proteins, and predict binding strength. We chose the top 1% of molecule-pocket pairs for further scoring, using refined binding calculations and machine-learning models to predict drug potency, drug distribution, and safety properties. We combined prediction scores to select about 120 molecules for laboratory tests against viral proteins and whole-virus surrogates. Of 91 compounds initially screened against the main protease, four inhibited protease activity by nearly 100% at the tested concentration. Of 32 compounds screened against the spike protein, two inhibited surrogate virus infection by greater than 50%. Three of four compounds tested against SARS-CoV-2 inhibited cell infection with good margins between effective and toxic concentrations. These molecules are promising candidates for optimization towards more effective and selective therapeutics.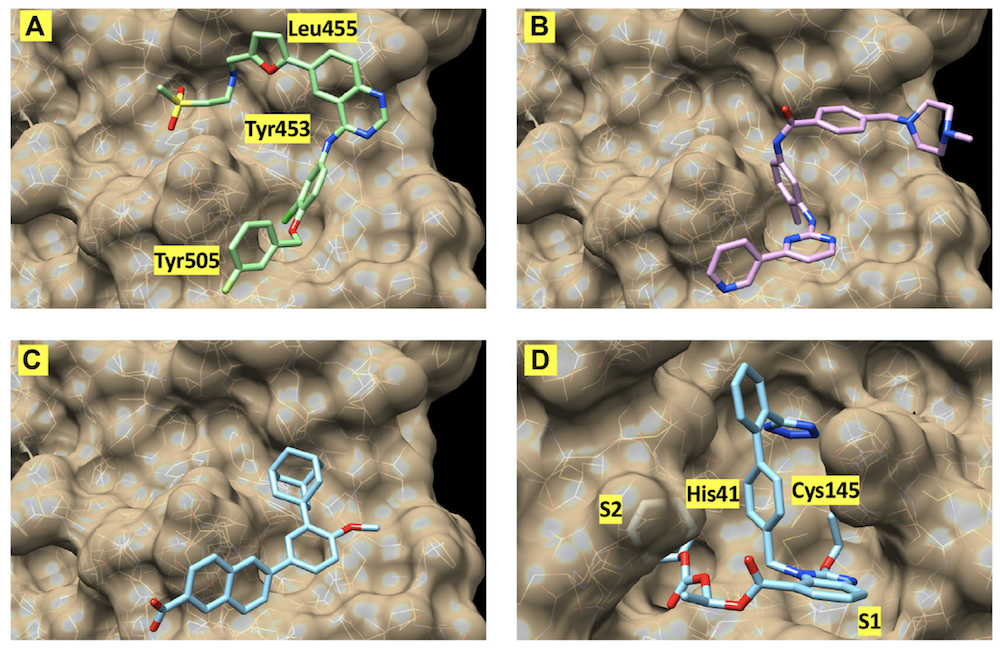
The images show the best-scoring docking pose for four of the inhibitors. The first three images show (A) lapatinib, (B) imatinib, and (C) adapalene, all docked to the receptor binding domain of the SARS-CoV-2 "spike" protein. (D) shows the best-scoring docking pose for candesartan cilexetil to the main protease of SARS-CoV-2. The labels identify protein residues neighboring the docked compounds.
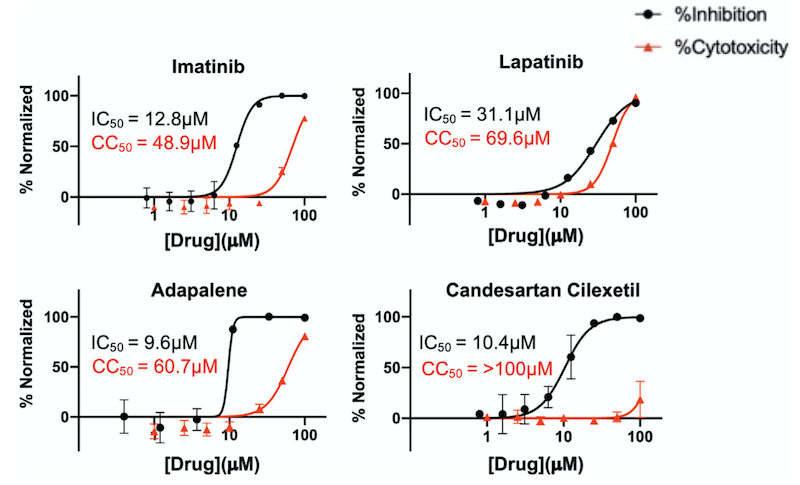
The graphs show percentage inhibition and percentage cytotoxicity from SARS-CoV-2 infection studies that show large therapeutic indexes in three hits. Varying concentrations of imatinib, lapatinib, and adapalene were used to treat virus for 30 min prior to infection in Vero cells (a commonly-used mammalian cell line). Candesartan cilexetil was added directly to cells without pre-treatment to virus. Data are normalized to percent inhibition or percent cytotoxicity for drug-treated cells vs. no-treatment control. The values are means, with error bars displaying standard deviation between triplicate tests. Half-maximal inhibitory concentration (IC50) curves and values are represented in black while half-maximal cytotoxicity concentration 50 (CC50) curves and values are represented in red.
See Discovery of Small-molecule Inhibitors of SARS-CoV-2 Proteins Using a Computational and Experimental Pipeline, Frontiers in Molecular Biosciences (2021) 8 644.
Research partnership with the American Heart Association
Contact: Felice Lightstone
BBS, along with machine-learning and database experts from the Global Security division and others at LLNL, is in a strategic business partnership with the American Heart Association to computationally predict interactions between drugs and human proteins. Such interactions are the basis of both drug effectiveness - do drugs "hit" their targets? - and adverse side effects - do drugs have deleterious effects on "off-target" proteins? Predictions will also be tested experimentally. A computational pipeline and a database of calculated interactions are being created.
In silico prediction of drug-protein binding is computationally expensive. Computational and experimental results are being used to train machine-learning models, which can make large-scale screening of novel drug candidates - even molecules that have yet to be synthesized - computationally feasible. This will assist in increasing the success rate of drug-discovery efforts and in accelerating drug development.
This collaboration is part of the AHA's newly formed Center for Accelerated Drug Discovery, an element of the AHA’s efforts to to improve people’s health by accelerating the pace at which drug-protein interactions are identified and documented before actual clinical trials begin.
It takes an average of 10 years for a new medicine to be commercialized in the marketplace and an average cost of $2.6 billion. As the population continues to age, the need for medications to treat chronic conditions, such as heart disease, Alzheimer’s disease, and diabetes, will continue to increase, challenging the current system to remove barriers to affordable health care and preventive medicine.
In this new partnership, AHA and LLNL aim to develop computational and experimental tools that will allow the biomedical community to validate targeted drug hypotheses that have higher probabilities of success and reduce time to market.
See also: Lawrence Livermore and American Heart Association partner to accelerate drug discovery
An electrostatic funnel in the GABA-binding pathway
Contact: Tim Carpenter
Neurotransmitters convey signals from one neuron to the next and are indispensable to the functioning of the nervous system. These small molecules bind to receptors to exert their action. One of the most important neurotransmitters is gamma-aminobutyric acid (GABA), which binds to its type A receptor (GABAA-receptor) to exert an inhibitory influence on the neuron. Many drugs, both medicinal and nefarious, bind to these GABAA-receptors and alter the balance of neuronal signals in the brain. There is a fine balance between these drugs eliciting the desired effect, and causing unwanted and sometimes irreversible alterations in neural behavior.
|
The average GABA binding pathway illustrates the narrowing of the standard deviation of GABA molecule positions as the molecule approaches the binding site. Each disk is centered at the average GABA position at that distance from the binding site (red circle). The disk's diameter is proportional to the standard deviation of the GABA position at that distance. The disks are oriented with their axes intersecting the binding site. The ligand-binding domain of the receptor is shown in gray. |
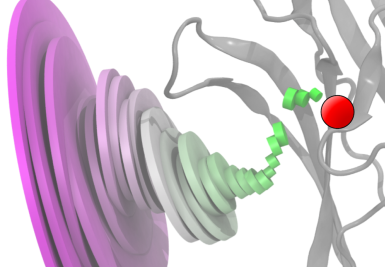
To study this critical binding event, we used many molecular dynamics computational simulations to observe precisely how the GABA molecule binds to GABAA-receptor. One hundred individual simulations were carried out where GABA was placed near the binding site and then allowed to freely bind to the GABAA-receptor. Binding occurred in 19 of these simulations. Statistical analysis of these binding simulations reveals the consistent electrostatics-driven pathway taken by GABA molecules to enter the binding site. Improved understanding of binding events enables the development of safer medicinal neuroactive drugs and countermeasures for effects of neuronal chemical trauma.
See An Electrostatic Funnel in the GABA-Binding Pathway Timothy S. Carpenter and Felice C. Lightstone PLoS Comput. Biol. (2016) 12(4): e1004831.
|
The average dipole experienced by GABA molecules as they follow the binding pathway indicates that the molecules are aligned in the protein’s electrostatic field. The orientation of the average dipole of the GABA molecules at that distance from the reaction site (red circle) is represented by an arrow, with the length of the arrow proportional to the strength of the average dipole. Blue-gray-red "filaments" show selected electric field-lines. |
Drug development using high-performance computing for a new class of antibiotics
Contact: Felice Lightstone
|
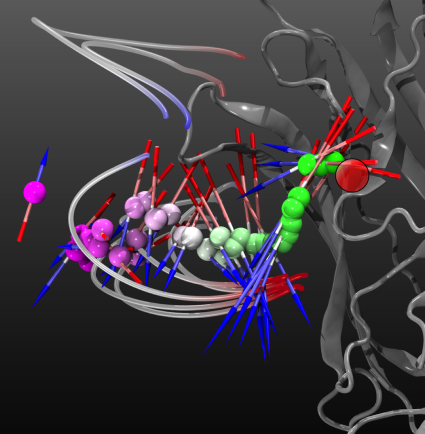
Figure 1. The initial fragment hit, bound to E. faecalis GyrB. The coloring of the GyrB enzyme indicates residue conservation across members of the GyrB evolutionary family; green regions are identical across members, yellow are conserved regions, and red are variable/non-conserved regions. |
The increasing spread of bacterial strains that are resistant to current antibiotic drugs is a serious problem. Structure-based drug design using high-performance computing has helped create the first new class of antibiotics in 30 years. This promising drug candidate has broad activity and effectiveness, and in addition, will hamper the evolution of resistance among its bacterial targets.
A number of important antibiotics target the bacterial topoisomerases DNA gyrase (GyrB) or topoisomerase IV (ParE), which are essential enzymes that control the topological state of DNA during replication. The effectiveness of these drugs, however, is being lost due to the spread of drug-resistant bacterial strains. While disabling either GyrB or ParE will kill the bacterium, a “dual-target” drug, which disables both enzymes, is less likely to succumb to drug resistance because two independent advantageous mutations would have to occur simultaneously in a bacterium in order for it to survive.
|
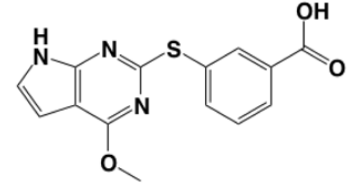
Figure 2. Chemical structure of initial fragment. |
|
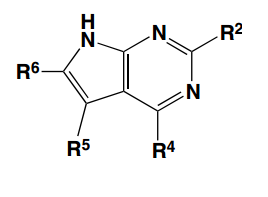
Figure 3. Scaffold of the molecule, with potential R groups labeled. |
Development of the candidate antibiotic began with the identification of a molecular fragment that weakly disrupts the activity of both the GyrB and ParE enzymes through its interaction with GyrB and ParE inhibitory binding pockets. The fragment binds to the pocket as Figure 1 shows with GyrB from E. faecalis.
The scaffold of this fragment has several R groups that can be modified or added; Figure 2 shows the chemical structure of the fragment, and Figure 3 shows the scaffold with R groups labeled. The R groups interact with the GyrB binding pocket in different ways, as Figure 4 shows.
|
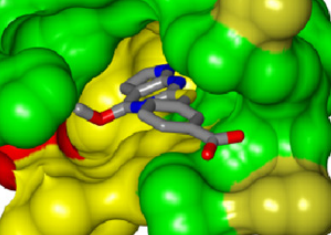
Figure 4. View of the binding pocket surface. The potential hydrogen-bonds between the molecule and conserved adenine-binding aspartate and structural water molecule are shown. The R5 and R6 groups face the active-site pocket interior, while the R2 and R4 groups are directed towards partially solvent-exposed faces of the active-site pocket. |
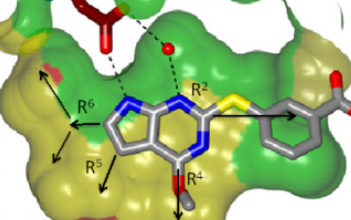
Potential R-group modifications were first explored with virtual screening, computational molecular dynamics simulations, and free-energy calculations, as well as quantum mechanical calculations, of the candidate drug molecules. Modifications that would produce increased affinity of the drug to GyrB and ParE were identified; the best of these were synthesized and tested in vitro.
High-performance computing enables the quick turnaround of high-quality, compute-intensive calculations. This was important because of the short development cycle – test, modify, test – involved in this effort.
Many, many alternatives were considered. Timely computer-based screening of options helps focus development efforts and helps to minimize costly wet-lab expenditures, such as drug synthesis.
Alternatives explored included substituents in three distinct regions of the active-site pocket: (i) the lipophilic active-site interior, along the R5 and R6 group vectors, (ii) the salt-bridge pocket, along the R2 group vector, and (iii) the residues and ordered solvent network at the mouth of the lipophilic pocket, along the R4 group vector. Filling the lipophilic pocket interior via installation of an ethyl group at R6 and a small substituent at R5 resulted in drug molecules with potencies two to three orders of magnitude greater than that of the initial fragment.
Particular attention was paid to optimizing the dual-targeting capabilities of the drug molecule, that is, affinity for both GyrB and ParE, while also minimizing potential off-target activity.
Thus, this drug development effort used high-performance computing to explore structure-based inhibitor design against multiple members of the target protein family, resulting in an inhibitor series with exquisite potency, broad enzymatic spectrum, and dual-targeting activity. The series is highly ligand-efficient, and tolerates significant chemical diversity at two R-group locations without compromising enzyme potency and spectrum. Taken together, these features greatly improve the prospects for developing molecules with antibacterial activity and good drug-like properties.
This work was done in collaboration with Trius Therapeutics, Inc., San Diego, CA
See L. W. Tari, X. Li, M. Trzoss, D. C. Bensen, Z. Chen, T. Lam, J. Zhang, S. J. Lee, G. Hough, D. Phillipson, S. Akers-Rodriguez, M. L. Cunningham, B. P. Kwan, K. J. Nelson, A. Castellano, J. B. Locke, V. Brown-Driver, T. M. Murphy, V. S. Ong, C. M. Pillar, D. L. Shinabarger, J. Nix, F. C. Lightstone, S. E. Wong, T. B. Nguyen, K. J. Shaw, J. Finn (2013) Tricyclic GyrB/ParE (TriBE) Inhibitors: A New Class of Broad-Spectrum Dual-Targeting Antibacterial Agents. PLoS ONE, 8(12): e84409.